Tutorial for spatial multi-omics vertical integration using DIRAC
by CHANG XU changxu@nus.edu.sg.
Last update: October 4th 2024
Installation
Make sure
sodiracis installed viapip install sodirac.The Python environment needs to install the packages listed in
requirements.txt.
[1]:
########## load packages
import os
import random
import pandas as pd
import numpy as np
import torch
import scanpy as sc
import matplotlib.pyplot as plt
######## load dirac
import sodirac as sd
2024-10-07 12:26:42.619665: I external/local_xla/xla/tsl/cuda/cudart_stub.cc:32] Could not find cuda drivers on your machine, GPU will not be used.
2024-10-07 12:26:44.578123: I external/local_xla/xla/tsl/cuda/cudart_stub.cc:32] Could not find cuda drivers on your machine, GPU will not be used.
2024-10-07 12:26:44.831249: E external/local_xla/xla/stream_executor/cuda/cuda_fft.cc:485] Unable to register cuFFT factory: Attempting to register factory for plugin cuFFT when one has already been registered
2024-10-07 12:26:45.405536: E external/local_xla/xla/stream_executor/cuda/cuda_dnn.cc:8454] Unable to register cuDNN factory: Attempting to register factory for plugin cuDNN when one has already been registered
2024-10-07 12:26:45.594850: E external/local_xla/xla/stream_executor/cuda/cuda_blas.cc:1452] Unable to register cuBLAS factory: Attempting to register factory for plugin cuBLAS when one has already been registered
2024-10-07 12:26:46.700173: I tensorflow/core/platform/cpu_feature_guard.cc:210] This TensorFlow binary is optimized to use available CPU instructions in performance-critical operations.
To enable the following instructions: AVX2 FMA, in other operations, rebuild TensorFlow with the appropriate compiler flags.
2024-10-07 12:26:55.688819: W tensorflow/compiler/tf2tensorrt/utils/py_utils.cc:38] TF-TRT Warning: Could not find TensorRT
[easydl] tensorflow not available!
Download the data
if you are have
wgetinstalled, you can run the following code to automatically download and unzip the data.
[ ]:
# Skip this cells if data are already download
! wget -O 0725_Protein.h5ad "https://drive.google.com/uc?export=download&id=1en6bitR0ROS6eh-RgNJECErVVAzLMiok"
! wget -O 0725_RNA.h5ad "https://drive.google.com/uc?export=download&id=1uclBRs1I4Q1Yvq0eUfZbBwPiOVgFaKMt"
if you do not have wget installed, manually download data from the links below:
DBiT-seq collected from ADT: https://drive.google.com/file/d/1en6bitR0ROS6eh-RgNJECErVVAzLMiok/view?usp=drive_link.
DBiT-seq collected from Protein: https://drive.google.com/file/d/1uclBRs1I4Q1Yvq0eUfZbBwPiOVgFaKMt/view?usp=drive_link.
[2]:
def seed_torch(seed=1029):
random.seed(seed)
os.environ['PYTHONHASHSEED'] = str(seed)
np.random.seed(seed)
torch.manual_seed(seed)
torch.cuda.manual_seed(seed)
torch.cuda.manual_seed_all(seed)
# if you are using multi-GPU.
torch.backends.cudnn.benchmark = False
torch.backends.cudnn.deterministic = True
seed_torch(seed=0)
[3]:
######### Define colors for spatial multi-omics domain
colormaps= ["#E41A1C", "#377EB8", "#4DAF4A", "#984EA3", "#FF7F00", "#A65628", "#FFFF33", "#F781BF", "#999999",
"#E5D8BD", "#B3CDE3", "#CCEBC5", "#FED9A6", "#FBB4AE", "#8DD3C7", "#BEBADA", "#80B1D3", "#B3DE69", "#FCCDE5",
"#BC80BD", "#FFED6F", "#8DA0CB", "#E78AC3", "#E5C494", "#CCCCCC", "#FB9A99", "#E31A1C", "#CAB2D6","#6A3D9A", "#B15928"]
Load DBiT-seq data
Here, DBiT-seq contains 936 spots, in each of which there are 22,802 genes and 22 proteins measured.
[4]:
data_path = "/home/project/11003054/changxu/Data/DBiT_seq"
data_name = "0725"
methods = "DIRAC"
[5]:
adata_RNA = sc.read(os.path.join(data_path, f"{data_name}_RNA.h5ad"))
adata_Protein = sc.read(os.path.join(data_path, f"{data_name}_Protein.h5ad"))
adata_RNA.raw = adata_RNA
adata_Protein.raw = adata_Protein
[6]:
adata_Protein[adata_RNA.obs_names]
[6]:
View of AnnData object with n_obs × n_vars = 936 × 22
obs: 'x', 'y', 'ground_truth'
obsm: 'spatial'
Prepare to Integrate
Following the workflow of single-cell preprocessing, we performed data cleaning and preparation. To accelerate model training, we applied PCA for dimensionality reduction.
We offer multiple methods for constructing spatial neighborhood graphs, including “knn,” “KDTree,” and “Radius.” Here, we chose the
KDTreemethod and set the number of nearest neighbors to 8.For the parameter k, extensive experiments indicate that values between 6 and 15 yield optimal results. This selection is also dependent on the resolution of the omics data being processed; generally, higher resolution data benefits from a larger number of spatial neighbors.
The
save_pathparameter should be specified. If not provided, the output will default to the current directory.
[7]:
######### Data processing
n_clusters = len(adata_RNA.obs["ground_truth"].unique())
adata_RNA.obs["Omics"] = data_name + "_mRNA"
adata_RNA.obs['Omics'] = adata_RNA.obs['Omics'].astype('category')
adata_Protein.obs["Omics"] = data_name + "_Protein"
adata_Protein.obs['Omics'] = adata_Protein.obs['Omics'].astype('category')
[8]:
######### Data processing
sc.pp.filter_genes(adata_RNA, min_cells=3)
sc.pp.normalize_total(adata_RNA)
sc.pp.log1p(adata_RNA)
sc.pp.highly_variable_genes(adata_RNA, n_top_genes=3000, subset=True, flavor='seurat_v3')
sc.pp.scale(adata_RNA)
sc.tl.pca(adata_RNA, n_comps=40)
[9]:
sc.pp.normalize_total(adata_Protein)
sc.pp.log1p(adata_Protein)
sc.pp.scale(adata_Protein)
[10]:
adj = graph(data = adata_RNA.obsm["spatial"],
rad_cutoff = 1.5,
k = 8,
distType= "KDTree", ### "BallTree", "Radius", "kneighbors_graph",'KDTree'
).main()["adj_norm"]
row, col, edge_attr = adj.t().coo()
edge_index = torch.stack([row, col], dim=0)
[11]:
y = pd.Categorical(
np.array(adata_RNA.obs["Omics"]),
categories=np.unique(adata_RNA.obs["Omics"]),
).codes
save_path = os.path.join("/home/users/nus/changxu/scratch/section1/Results", f"{data_name}_{methods}")
if not os.path.exists(save_path):
os.makedirs(save_path)
Training DIRAC
[12]:
###### Training Dirac for spatial multi-omics
dirac = sd.integrate_app(save_path = save_path, use_gpu = True, subgraph = False)
samples = dirac._get_data(dataset_list = [adata_RNA.obsm["X_pca"].copy(), adata_Protein.X.copy()],
batch_list = [y, y],
domain_list = [np.zeros(adata_RNA.shape[0]), np.ones(adata_Protein.shape[0])],
edge_index = edge_index,
num_parts = 10,
num_workers = 1,)
models = dirac._get_model(samples,
n_hiddens = 128,
n_outputs = 64,
opt_GNN = "GCN",)
data_z, combine_recon, now = dirac._train_dirac_integrate(samples = samples,
models = models,
epochs = 500,
lamb = 5e-4,
scale_loss = 0.025,
lr = 1e-3,
wd = 5e-2,)
Found 2 unique domains.
Project..: 100%|█| 500/500 [00:12<00:00, 40.36it/s, Loss=73.3, D
[13]:
adata_RNA.obsm[f"{methods}_embed"] = data_z[: adata_RNA.shape[0], :]
adata_Protein.obsm[f"{methods}_embed"] = data_z[adata_RNA.shape[0] :, :]
adata_RNA.obsm["combine_recon"] = combine_recon
adata_Protein.obsm["combine_recon"] = combine_recon
[16]:
####### Combined
sc.pp.neighbors(adata_RNA, use_rep='combine_recon')
res_RNA_ATAC = _priori_cluster(adata_RNA, eval_cluster_n = n_clusters)
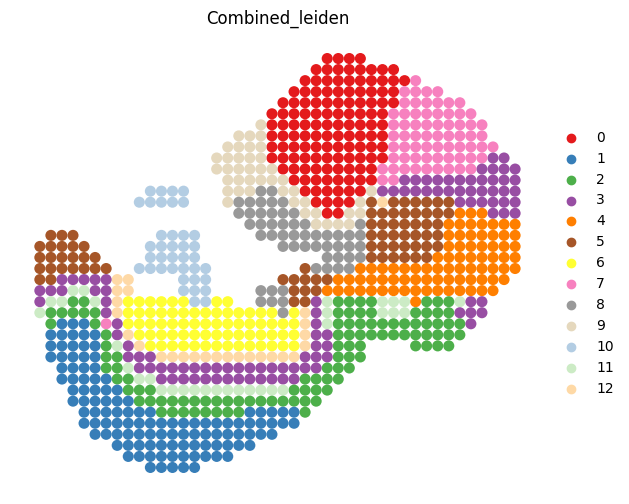
sc.tl.leiden(adata_RNA, resolution = res_RNA_ATAC, key_added="Combined_leiden")
# mclust_R(adata=adata_RNA, num_cluster = n_clusters, used_obsm="combine_recon",key_added="Combined_mclust")
# sc.pl.spatial(adata_RNA, color=["Combined_mclust", "Combined_leiden", "ground_truth"], palette = colormaps, frameon=False, spot_size=1)
fig, ax = plt.subplots(figsize=(8, 6))
sc.pl.spatial(adata_RNA, color=["Combined_leiden"], palette = colormaps, frameon=False, spot_size=1, ax=ax)
fig.savefig(os.path.join(save_path, f"{data_name}_{methods}_Combined_spatial_{now}.pdf"), bbox_inches='tight', dpi = 300)
Save data
[18]:
adata_RNA.write(os.path.join(save_path, f"{data_name}_{methods}_RNA_Final_{now}.h5ad"), compression="gzip")
adata_Protein.write(os.path.join(save_path, f"{data_name}_{methods}_Protein_Final_{now}.h5ad"), compression="gzip)